
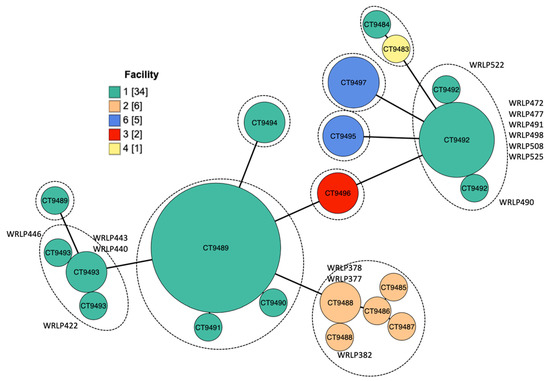
Recent listeriosis outbreaks linked to fresh produce suggest the need to better understand and mitigate L. monocytogenes contamination in packing and processing environments. Using whole genome sequencing (WGS) and phenotype screening assays for sanitizer tolerance, we characterized 48 L. monocytogenes isolates previously recovered from environmental samples in five produce handling facilities. Within the studied population there were 10 sequence types (STs) and 16 cgMLST types (CTs). Pairwise single nucleotide polymorphisms (SNPs) ranged from 0 to 3047 SNPs within a CT, revealing closely and distantly related isolates indicative of both sporadic and continuous contamination events within the facility. Within Facility 1, we identified a closely related cluster (0–2 SNPs) of isolates belonging to clonal complex 37 (CC37; CT9492), with isolates recovered during sampling events 1-year apart and in various locations inside and outside the facility. The accessory genome of these CC37 isolates varied from 94 to 210 genes. Notable genetic elements and mutations amongst the isolates included the bcrABC cassette (2/48), associated with QAC tolerance; mutations in the actA gene on the Listeria pathogenicity island (LIPI) 1 (20/48); presence of LIPI-3 (21/48) and LIPI-4 (23/48). This work highlights the potential use of WGS in tracing the pathogen within a facility and understanding properties of L. monocytogenes in produce settings. View Full-Text


